Klinische Bedeutung
Motoneuronerkrankungen (MND) sind eine Gruppe neurodegenerativer Erkrankungen, die durch Degeneration der oberen und unteren Motoneurone charakterisiert sind. Zur Gruppe der MND werden unter anderem die amyotrophe Lateralsklerose (ALS), die primäre Lateralsklerose, die progressive Muskelatrophie und die Pseudobulbärparalyse gezählt. Mit einer Inzidenz von etwa 2/100.000 Personen pro Jahr weltweit ist ALS die häufigste MND.
ALS-Patienten erleben in der frühen Phase des Krankheitsverlaufs zunächst unspezifische Symptome wie Muskelschwäche oder Krämpfe in Armen und Beiden bzw. Schluck- und Sprachbeschwerden. Im weiteren Krankheitsverlauf breiten sich die Beschwerden über den gesamten Körper aus und nehmen stetig an Stärke zu bis es schließlich zum Verlust der Selbstständigkeit und des Kommunikationsvermögens der Patienten kommt.
ALS ist nicht heilbar und der Großteil der Patienten erliegt der Krankheit 3 bis 5 Jahre nach Einsetzen der ersten Symptome. Während Mutationen in verschiedenen Genen gefunden wurden, die lediglich den kleinen Teil (5 bis 10 % aller ALS-Fälle) der familiären ALS-Fälle erklären, ist die grundlegende Ursache von sporadischer ALS trotz intensiver Forschung unbekannt.
Diagnose
Die El-Escorial-Kriterien zur Diagnose von ALS basieren auf der Beurteilung klinischer Symptome und der Beschreibung der Beeinträchtigung des unteren Motoneurons, z. B. anhand von Elektromyographie (EMG). Diese diagnostischen Kriterien sind zwar standardisiert, greifen jedoch erst zu einem späten Zeitpunkt in der Erkrankung, zu dem bereits eine Vielzahl der Motoneuronen degeneriert ist. Zwischen der ersten Beschreibung der Symptome und der Diagnose liegen oft mehr als 12 Monate, was insbesondere hinsichtlich der kurzen Lebenserwartung der Erkrankten fatal ist. Für eine schnellere Diagnose und die sichere Differenzierung von ALS gegenüber anderen Krankheiten, die insbesondere in den Anfangsstadien eine ALS-ähnliche Symptomatik aufweisen (MND-Mimics: Polyneuropathie, Myopathie, Sporadische Einschlusskörpermyositis, etc.) werden dringend neue diagnostische Methoden benötigt.
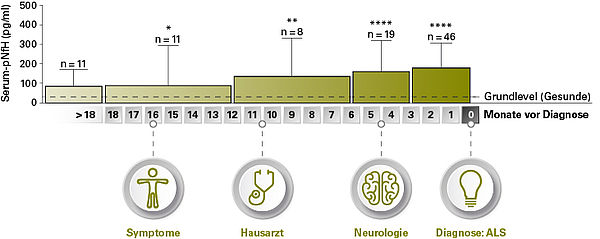 Ein vielversprechender Biomarker für ALS ist die phosphorylierte schwere Kette des Neurofilaments (phosphorylated Neurofilament heavy chain, pNf-H), die ein Hauptbestandteil des neuronalen Cytoskeletts ist und bei neuronaler Schädigung freigesetzt wird. Die Konzentration von pNf-H im Liquor von ALS-Patienten ist bereits im frühen Krankheitsstadium deutlich erhöht. Darüber hinaus korreliert die pNf-H-Konzentration mit dem Grad der Degeneration der Motoneuronen. Mehrere Studien legen nahe, dass die pNf-H-Werte in Liquor ein besseres Diagnostikum für ALS sind als die Werte der Neurofilament Leichtkette (NfL). Die pNf-H-Konzentration im Blut korreliert mit der Liquor-Konzentration und ist bereits bis zu 18 Monate vor der Diagnosestellung erhöht. Die pNf-H-Messung eignet sich somit zur Diagnostik, Differenzialdiagnostik und Prognose bei ALS. Es wird empfohlen, diesen Marker in die MND-Routinediagnostik aufzunehmen.
Ein vielversprechender Biomarker für ALS ist die phosphorylierte schwere Kette des Neurofilaments (phosphorylated Neurofilament heavy chain, pNf-H), die ein Hauptbestandteil des neuronalen Cytoskeletts ist und bei neuronaler Schädigung freigesetzt wird. Die Konzentration von pNf-H im Liquor von ALS-Patienten ist bereits im frühen Krankheitsstadium deutlich erhöht. Darüber hinaus korreliert die pNf-H-Konzentration mit dem Grad der Degeneration der Motoneuronen. Mehrere Studien legen nahe, dass die pNf-H-Werte in Liquor ein besseres Diagnostikum für ALS sind als die Werte der Neurofilament Leichtkette (NfL). Die pNf-H-Konzentration im Blut korreliert mit der Liquor-Konzentration und ist bereits bis zu 18 Monate vor der Diagnosestellung erhöht. Die pNf-H-Messung eignet sich somit zur Diagnostik, Differenzialdiagnostik und Prognose bei ALS. Es wird empfohlen, diesen Marker in die MND-Routinediagnostik aufzunehmen.
Brochüre: Neurofilamente in der Diagnostik - Biomarker für neuroaxonale Schädigungen


















